Protein aggregation in neurodegeneration
Lead researcher: Dr Han-Jou Chen, Department of Biology
As we go into the 21st century and with increased life expectancy, neurodegenerative disorders are affecting more people causing significant impact on individuals, families and society. As the underlying mechanisms are yet to be fully illustrated, currently there is no effective treatment for those diseases. To address this dire need for new understanding and new therapeutic strategy for neurodegeneration, our lab focuses on 1) the molecular chaperone system, and 2) the involvement of RNA regulation in neurodegeneration.
1. The Molecular Chaperone system
The majority of these diseases feature by the presence of aberrant protein aggregates in affected neuronal tissues which convey toxicity resulting in neuronal death and disease development [1].
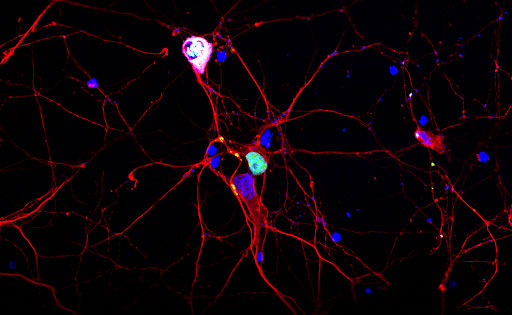
Figure 1. TDP-43 forms aggregates when over-expressed in neurones. TDP-43 (green) is a nuclear (DAPI in blue) localisation protein. However, when over-expressed in primary neurones (beta-3-tubulin, red), they form cytoplasmic aggregates which are hyperphosphorylated (magenta).
Our research aims to study the factors contributing to these aberrant protein aggregations and explore the potential of protein chaperone system activation for new therapeutic development. We found that the levels of several key components of this chaperone system are decreased in patients suffering from one of the neurodegenerative diseases, Amyotrophic Lateral Sclerosis (ALS) [2], and also our animal model of the disease [2,3].
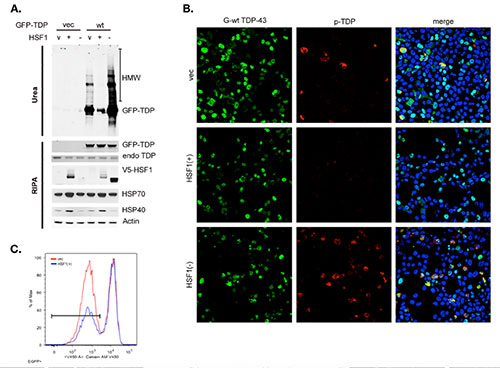
Figure 2. Activation of chaperone system by the expression of HSF1(+) rescues TDP-43 protein solubility (A), hyperphosphorylation (B) and toxicity (C).
Forced activation of this chaperone system efficiently reverses protein aggregation and enhances cell survival in petri dishes [2]. We are currently investigating the gene therapy approach to activate this system in animal models, and developing drug screening for pharmacological introversion.
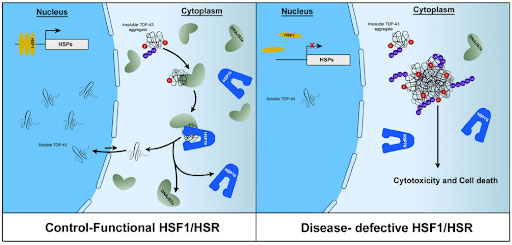
Figure 3. Model of the involvement of chaperone system in TDP-43 folding and proteinopathy. In the healthy condition with functional chaperone system, chaperone proteins HSP70 and DNJB2a recognise misfolded TDP-43, interact with it and return it to its native soluble state, which enables TDP-43 to shuttle back to the nucleus. In the diseased condition, this chaperone system is impaired by either reduced protein levels or decreased activity of HSF1, which subsequently fail to efficiently engage misfolded TDP-43. As a result, the insoluble and phosphorylated TDP-43 protein accumulate and form cytosolic aggregates which eventually lead to cytotoxicity and neuronal death.
2. RNA regulation
Recent studies have shown that various RNA-binding proteins are associated with a wide range of neurodegenerative diseases, indicating misregulation of RNA in neurodegeneration. Our lab is the first to characterise disease-associated mutations in TDP-43 that disrupt the interaction between TDP-43 protein and its target RNA [4]. Although mutations like these are rare in the patient population, wider research indicates that TDP-43-mediated RNA regulation is affected in other ALS patients and animal models not carrying an RNA-binding deficient mutation [5], suggesting the influence of TDP-43-meidated RNA misregulation in general neurodegeneration. To further illustrate the extent of involvement and precise pathogenesis mechanism it invokes, our lab is currently working on research projects characterising RNA-binding deficient TDP-43 in cell and animal models.
References
[1] Soto C and Estrada LD. (2008) Protein misfiling and Neurodegeneration. Arch Neurol 65(2): 184-9
[2] Chen et al (2016) The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain 139:1417-1432
[3] Mitchell et al (2015) Wild type human TDP-43 potentiates ALS-linked mutant TDP-43driven progressive motor and cortical neutron degeneration with pathological features of ALS. Acta Neuropathol Commun 3:36
[4] Chen et al (2019) RRM adjacent TARDBP mutations disrupt RNA binding and enhance TDP-43 proteinopathy. Brain 142(12):3753-3770
[5] Yu et al (2021) HSP70 chaperones RNA-free TDP-43 into anisotrophic intranuclear liquid shericl shells. Science 371(6529): eabb4309