Professor Jason Lynam
01904 322534
Email: jason.lynam@york.ac.uk
Transition Metal Organometallic Chemistry
New Methods to Probe Catalysis using Time-Resolved Infra-Red Spectroscopy
Catalysis plays a vitally important role in chemical synthesis and the development of new catalysts is enabled by knowing the mechanistic pathways which underpin their activity. However, observing these pathways is extremely difficult as they are often rapid and form a small component of a complex mixture. We have developed a method to circumvent these problems using time-resolved infra-red spectroscopy in collaboration with the ULTRA facility at the Rutherford Appleton Laboratory and Professor Ian Fairlamb.
Central to the developments is that light is used to activate a manganese carbonyl catalyst, simulating the pathways it undergoes in a thermal reaction. The light is provided by a laser pulse which is followed a short-time later by a second photon that interrogates the changes to the structure of the catalyst. The spectrometer at ULTRA allows us to observe the steps in reactions that occur from a picosecond (10-12 s) to a millisecond (10-3 s) so that even the fastest chemical process can be observed.
Using this method we have been able to observed key steps in manganese catalysed C-H functionalisation reactions - in the example shown below we are able to observe an alkyne binding to the metal followed by a the C-C bond formation occurring. In addition, we have used this method to understand the interactions between metal complexes and solvent molecules - an overlooked component of a reaction. Remarkably, our findings indicate that solvents such as tetrahydrofuran initially bind to the metal through the two electrons of a C-H bond before isomerisating to the more thermodynamiccaly favoured O-bound form.
Lead References L. Anders Hammarback, Ian P. Clark, Igor V. Sazanovich, Michael Towrie, Alan Robinson, Francis Clarke, Stephanie Meyer, Ian J. S. Fairlamb and Jason M. Lynam, Nature Catal., 2018, 1, 830. for associated commentary to this article see Congyang Wang Nature Catal., 2018, 1, 816-817. Benjamin J. Aucott, Anne-Kathrin Duhme-Klair, Benjamin E. Moulton, Ian P. Clark, Igor V. Sazanovich, Michael Towrie, L. Anders Hammarback, Ian J. S. Fairlamb and Jason M. Lynam, Organometallics 2019, 38, 2391. L. Anders Hammarback, Benjamin J. Aucott, Joshua T. W. Bray, Ian P. Clark, Michael Towrie, Alan Robinson, Ian J. S. Fairlamb and Jason M. Lynam, J. Am. Chem. Soc., 2021, 143, 1356. L. Anders Hammarback, Jonathan B. Eastwood, Thomas J. Burden, Callum J. Pearce, Ian P. Clark, Michael Towrie, Alan Robinson, Ian J. S. Fairlamb and Jason M. Lynam, Chem. Sci., 2022, 13, 9902.
Outer-sphere Electrophilic Fluorination of Organometallic Compounds
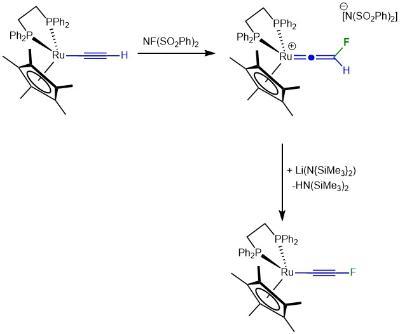
The formation of carbon-fluorine bonds is crucial for the synthesis of a vast number of pharmaceuticals and agrochemicals. Given that there are very few naturally occurring compounds with C-F bonds, highly selective and simple methods are required to introduce fluorine atoms into organic molecules are needed. In collaboration with Dr John Slattery we have developed an entirely new mechanistc pathway to achieve this goal. As shown below, the reaction of simple half-sandwhich ruthenium complexes with electrophilic fluorinainte agents results in direct C-F bond formation, circumventing the formation of metal-fluorine bonds which often occurs in related transition metal-catalysed reactions.
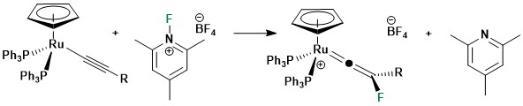
This has, in turn, permitted us to develop the synthetic of the first stable fluorinated alkyne in which the ruthenium essentially acts as sterically demanind protecting group.
Lead References Lucy M. Milner, Natalie E. Pridmore, Adrian C. Whitwood, Jason M. Lynam and John M. Slattery, J. Am. Chem. Soc. 2015, 137, 10753; Lucy M. Milner, Lewis M. Hall, Natalie E. Pridmore, Matthew K. Skeats, Adrian C. Whitwood, Jason M. Lynam and John M. Slattery, Dalton Trans., 2016, 45, 1717, Lewis M. Hall, David P. Tew, Natalie E. Pridmore, Adrian C. Whitwood, Jason M. Lynam, John M. Slattery, Angew. Chemie Int. Ed. 2017, 56, 7551.
New Catalysts for the Activation of Pyridine using Pyridylidene Intermediates
We have developed new methods for the direct C-H functionalisation of pyridine and its derivatives. Using half-sandwich ruthenium complexes, such as 1 we have demonstrated that it is possible to formally insert an alkyne into the C-H bond of pydridine (Scheme 1).1 The key mechanistic step in this process revolves around the formation of a remarkable class of ligands, pyridylidenes, which are an interesting class of metal carbene complexes. We have been able to isolate complexes containing these ligands (2) and (in collaboration with Dr John Slattery) used density functional theory to show that pyridylidene ligands are indeed key intermediates in the C-H bond activation and C-C bond formation steps.
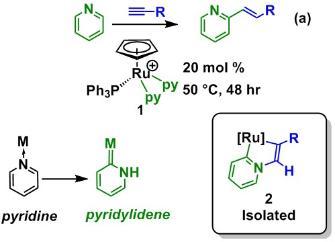
Furthermore, this study has allowed us to develop a one-pot protocol for this reaction which uses a commercially available ruthenium precursor and has allowed for facile evaluation of the factor affecting the efficiency of this reaction.
Lead References David G Johnson, Jason M Lynam, Neetisha S Mistry, John M Slattery, Robert J Thatcher, and Adrian C Whitwood, J. Am. Chem. Soc., 2013, 135, 2222. Jason M. Lynam, Lucy M. Milner, Neetisha S. Mistry, John M. Slattery, Sally R. Warrington and Adrian C. Whitwood, Dalton Trans., 2014, 43, 4565
Transition Metal Vinylidene Chemistry
A further area of research is focused around the organometallic chemistry of the transition metal elements and in particular, the understanding of the chemistry of alkynes within the coordination sphere of metals. The overall aim of this research is to utilise the reactivity of transition metal complexes to perform complex organic transformation in a selective manner. One example of this research is shown in Scheme 2. We have examined the conversion of the rhodium alkyne complex 3 into the resulting alkynyl hydride 4 and ultimately the vinylidene complex 5. Importantly, we were able to perform a kinetic analysis on the data (Figure 2) and demonstrate that 3 and 4 are in equilibrium and the formation of 5 is essentially irreversible. Central to this approach is a collaboration with Dr John Slattery in which the synergic relationship between experimental and theoretical studies may be used to gain valuable mechanistic insight.
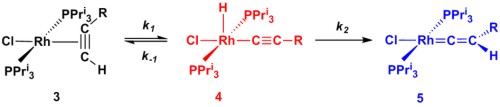
Scheme 2
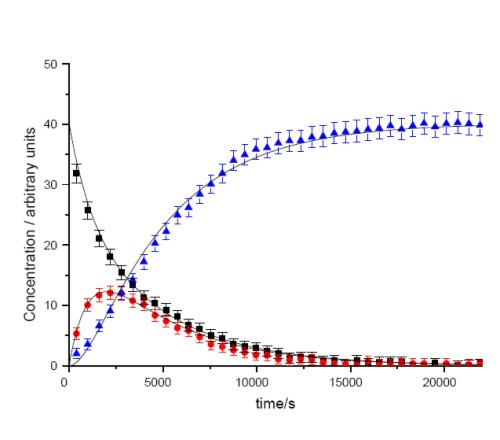
Figure 2
We have also investigated the role which a coordinate acetate ligand may affect the chemistry of transition metal alkyne and vinylidene ligand. A study of the system based on [Ru(κ2-OAc)2(PPh3)2] demonstrated that the reaction with alkynes proceeds far more rapidly than the corresponding chloride derivatives. The princple reason for the acceleration is the coordinated acetate ligand acting to both deprotonate and repronate the coordinated alkyne. This Ligand-Assisted Proton Shuttle (LAPS) process signficnaly lowers the barrier to the formation of the vinylidene complex (Figure 3). The [Ru(κ2-OAc)2(PPh3)2] fragment is also an excellent reporter for the electronic properties of a number of ligands and we have employed the changes in the spectroscopic and metric properties of this complex to evaluate the donor/acceptor properties of a range of ligands.6
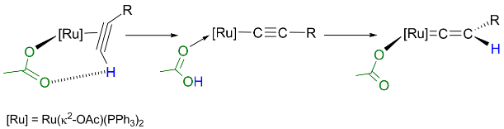
Scheme 3
In collaboration with Dr Natalie Fey (University of Bristol) we have also demonstrated how it is possible to use computational methods to evaluate the key factors involved in the stabilisation of transition metal vinylidene compelxes when compared to their alkyne tautomers. By calculating the structures and energies of a wide number of different metal complexes, alkynes and different ligand combinations it is possible to extrapolate the key factors needed to stabilise each tautomer.
Lead References Bernhard Breit, Urs Gellrich, Timothy Li, Jason M. Lynam, Lucy M. Milner, Natalie E. Pridmore, John M. Slattery and Adrian C. Whitwood, Dalton Trans., 2014, 43, 11277. Oliver J. S. Pickup , Iman Khazal , Elizabeth J. Smith , Adrian C. Whitwood , and Jason M. Lynam, Keshan Bolaky , Timothy C. King, Benjamin W. Rawe, and Natalie Fey Organometallics, 2014, 33, 1751.

