Dr John Slattery
01904 322610
E-mail: john.slattery@york.ac.uk
ORCiD: 0000-0001-6491-8302
ResearcherID: E-5578-2010
Experimental and computational investigations into ionic liquids and organometallic catalysts
We are interested in synthetic, structural and mechanistic investigations that focus predominantly on two areas: ionic liquids (ILs) and organometallic chemistry. These studies aim to provide new fundamental insights that underpin developments in applications such as homogeneous catalysis, organic synthesis, alternative solvent systems or novel electrolytes for batteries, solar cells or supercapacitors. One of our key interests is in mechanistic studies that aim to understand the mode of action of organometallic catalysts with a view to improving their activity.
Our work involves many different aspects of chemistry including synthesis (e.g. of ILs, organometallic complexes and main-group compounds), fundamental studies of ILs (e.g. physical property measurements, X-ray and neutron scattering studies), organometallic chemistry, computational chemistry and catalysis. We are part of the Inorganic group in the Department of Chemistry, but our projects also frequently involve organic chemistry, materials characterisation, quantum chemistry and physical chemistry approaches.
We are always interested to hear from enthusiastic and talented students and post-doctoral researchers looking to join the group (for PhD projects, post-doctoral positions, MChem research projects, Erasmus placements etc.) Please contact John Slattery (john.slattery@york.ac.uk) to discuss currently available projects.
Ionic liquids
Ionic liquids (ILs) are an interesting class of salts with surprisingly low melting points. They are often liquid at room temperature, which is remarkable considering the high melting points of classical inorganic salts such as NaCl (m.p. 801°C). ILs are usually composed of a relatively large organic cation in combination with a complex anion e.g. [C2mim][Tf2N] or [C12mim][Tf2N] (below).
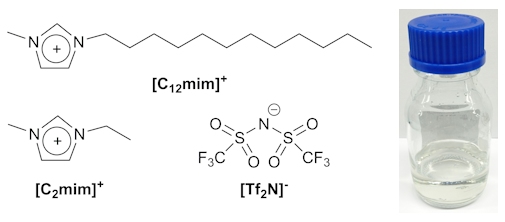
The structures of some common IL ions (left) and a sample of [C2mim][Tf2N] (right).
ILs are currently attracting considerable attention, as their unique properties make them suitable for a wide range of applications including as “green” solvents; electrolytes for batteries, solar cells and supercapacitors; in biomass processing and many more. We have worked on a range of projects involving ILs. These include building predictive models to aid in IL design, the development of task-specific ILs for catalysis (in collaboration with Fairlamb, York), probing the surface structure of ILs (in collaboration with McKendrick, Heriot-Watt and Minton, Montana State) and investigations into the surface and bulk structure of IL mixtures. We are also involved in a collaboration with Duncan Bruce (York) and José Nuno Canongia Lopes (IST, Lisbon) investigating the synthesis of new ionic liquid crystals (ILCs) and their use as structured reaction media to control the rate and/or stereochemical outcome of pericyclic organic reactions.
Organometallic chemistry and catalysis
Organometallic chemistry underpins a large proportion of homogeneous catalysis, which is crucial in academia and industry to carry out reactions with greater efficiency under milder conditions. It is essential for the production of pharmaceuticals, agrochemicals and plastics and plays a critical role in sustainable chemistry. We are interested in developing novel organometallic processes that could form the basis of new catalytic reactions and in understanding the mechanistic features of existing catalysts in order to provide insights that can be used to improve their efficiency and/or selectivity.
As part of an EPSRC-funded project, and a long-running collaboration with Lynam (York), we investigated the mechanism of a ruthenium-catalysed pyridine alkenylation reaction (below). This work found that unusual carbene ligands (pyridylidenes) play an unexpected role in the catalytic cycle. This mechanistic understanding enabled significantly improved catalysts to be developed.
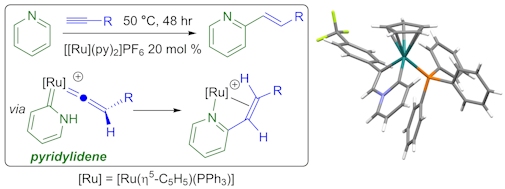
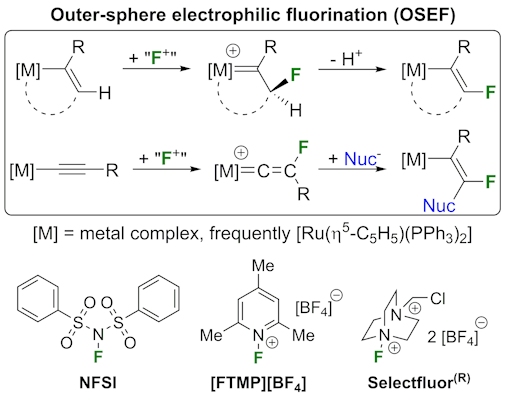
Also in collaboration with Lynam, we discovered an entirely new way for metals to facilitate the formation of C-F (or C-CF3) bonds, which we call outer-sphere electrophilic fluorination (OSEF). In OSEF a metal alkenyl or alkynyl complex reacts with an electrophilic fluorinating agent (e.g. Selectfluor®) to directly form a new C-F bond. OSEF reactions occur rapidly, under mild conditions and with complete regio- and diastereo-selectivity. This has provided us with a selective and versatile route to prepare unusual fluorinated organometallic complexes, for example the first structurally characterised fluoroalkyne.
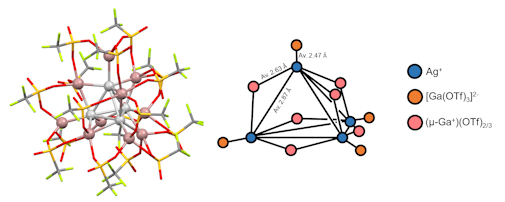
We also have an interest in main-group-element organometallics and have recently explored the complex speciation in a promising new gallium-based catalyst for C-C bond forming reactions - "gallium triflate". It was found that this compound contains both cationic and anionic gallium species, which display a range of oxidation states. The nature of the salts isolated from solutions of "gallium triflate" in aromatic solvents depends on both the solvent and the presence of ligands, such as 18-crown-6. In one case, a salt containing a dianionic Ag/Ga/triflate cluster (below), presumed to be an intermediate in the formation of "gallium triflate" was observed.
Selected recent publications
- Insights into the composition and structural chemistry of gallium(I) triflate
Joseph Boronski, Matthew Stevens, Bono van IJzendoorn, Adrian C. Whitwood and John M. Slattery, Angew. Chem. Int. Ed., 2021, 60, 1567-1572. - Probing Conformational Heterogeneity at the Ionic Liquid-Vacuum Interface by Reactive-Atom Scattering
Eric J. Smoll Jr., Simon M. Purcell, Lucia D’Andrea, John M. Slattery, Duncan W. Bruce, Matthew L. Costen, Kenneth G. McKendrick, and Timothy K. Minton, J. Phys. Chem. Lett., 2019, 10, 156–163. - Filling a Niche in “Ligand Space” with Bulky, Electron‐Poor Phosphorus(III) Alkoxides
Sharifa Hussein, Denis Priester, Paul Beet, Jonathon Cottom, Sam J. Hart, Tim James, Robert J. Thatcher, Adrian C. Whitwood and John M. Slattery, Chem. Eur. J., 2019, 25, 2262-2271. - A structurally characterised fluoroalkyne
Lewis M. Hall, David P. Tew, Natalie E. Pridmore, Adrian C. Whitwood, Jason M. Lynam and John M. Slattery, Angew. Chem. Int. Ed., 2017, 56, 7551-7556. - Nano-Segregation and Structuring in the Bulk and at the Surface of Ionic-Liquid Mixtures
Duncan W. Bruce, Christopher P. Cabry, José N. Canongia Lopes, Matthew L. Costen, Lucía D’Andrea, Isabelle Grillo, Brooks C. Marshall, Kenneth G. McKendrick, Timothy K. Minton, Simon M. Purcell, Sarah Rogers, John M. Slattery, Karina Shimizu, Eric Smoll, and María A. Tesa-Serrate, J. Phys. Chem. B, 2017, 121, 6002-6020. - Liquid-Crystalline Ionic Liquids as Ordered Reaction Media for the Diels–Alder Reaction
Duncan W. Bruce, Yanan Gao, José Nuno Canongia Lopes, Karina Shimizu and John M. Slattery, Chem. Eur. J., 2016, 22, 16113-16123. - Outer-sphere electrophilic fluorination of organometallic complexes
Lucy M. Milner, Natalie E. Pridmore, Adrian C. Whitwood, Jason M. Lynam, and John M. Slattery, J. Am. Chem. Soc., 2015, 137, 10753-10759. - Access to novel fluorovinylidene ligands via exploitation of outer-sphere electrophilic fluorination: new insights into C–F bond formation and activation
Lucy M. Milner, Lewis M. Hall, Natalie E. Pridmore, Matthew K. Skeats, Adrian C. Whitwood, Jason M. Lynam and John M. Slattery, Dalton Trans., 2016, 45, 1717-1726. - Structure of Amido-Pyridinium Betaines: Persistent Intermolecular C-H•••N Hydrogen Bonding in Solution
Robert J. Thatcher, David G. Johnson, John M. Slattery and Richard E. Douthwaite, Chem. Eur. J., 2016, 22, 3414-3421. - Ruthenium-Mediated C−H Functionalization of Pyridine: The Role of Vinylidene and Pyridylidene Ligands
David G. Johnson, Jason M. Lynam, Neetisha S. Mistry, John M. Slattery, Robert J. Thatcher, and Adrian C. Whitwood, J. Am. Chem. Soc., 2013, 135, 2222−2234.

