Department of Biology
Proteomics & Biological Mass Spectrometry Case Studies
Proteomics
Expression Proteomics
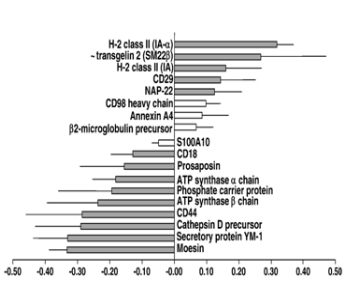
Protein quantification using stable isotope labels is a central technology in modern proteomics. We used isobaric tags for relative and absolute quantitation (iTRAQ) (ref) to look at changes in protein expression levels in dendritic cells. The study pointed to differing metabolic states of dendritic cells mediating different types of lymphocyte-mediated immunity (ref).
 |
 |
Protein Quantification
Protein Quantification
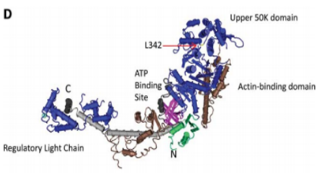
Using mass spectrometry for protein quantification allows specific sequences to be targeted and adding stable isotope-labelled surrogates allows protein levels to be measured without the use of antibodies (ref, ref). We recently applied this technique to determine the amount of a mutated myosin (MYH4L342Q) in mouse muscle. The ability to provide a direct and specific measurement of the level of mutated protein was crucial to publication of the study (ref), which revealed the underlying mechanism of a myofibrillar myopathy.
 |
 |
PTMs
Post-Translational Modifications
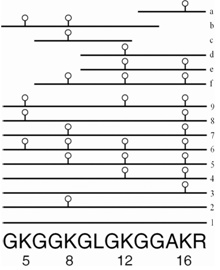
Mass spectrometry is arguably the most powerful tool available for identifying, locating, and quantifying post-translational modifications of proteins (ref). We were able to identify multiple sites of acetylation and methylation of plant histones H3 and H4. The study (ref) revealed previously unseen patterns of histone acetylation in Arabidopsis following X-ray treatment suggesting a role in the genotoxic stress response.
 |
 |
Metabolite Quant
Metabolite Quantification
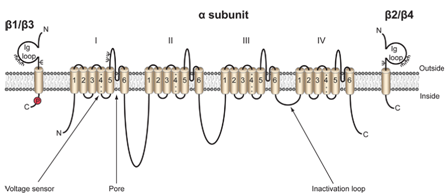
Multiple-reaction monitoring (MRM) is a mass spectrometry method that provides exquisite sensitivity and selectivity for the quantification of small molecules, and is an indispensable tool in drug metabolism, pharmacokinetics, and toxicology (ref). We have developed an MRM-based assay for the anticonvulsant phenytoin, which blocks voltage-gated sodium channels. The assay is being in studies of the use of phenytoin as a treatment for breast cancer (ref, ref).
 |
 |
Ligand ID
Ligand Identification
Identifying unknown compounds relies heavily on analytical techniques such as mass spectrometry. We recently took advantage of the ultra-high mass accuracy of Fourier-transform, ion cyclotron resonance mass spectrometry (FT-ICR-MS) to identify a novel cofactor in the putative tRNA-modifying enzyme CmoA. The results of the study (ref) suggest that S-adenosyl-L-methionine (SAM) might not always be the methyl group donor in DNA and RNA methylation.
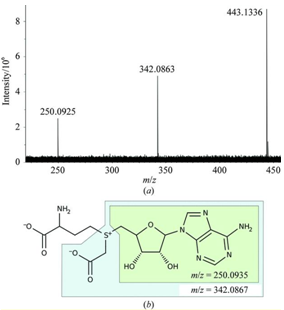 |
|

Glycobiology
Glycobiology
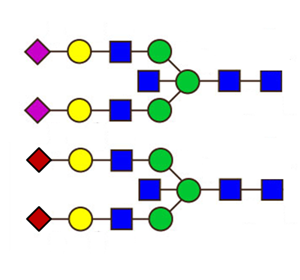
Glycosylation plays an important role in protein function and the re-modelling of glycans is a way to increase therapeutic efficacy. We have applied an analytical method, which was developed in‑house (ref) to characterise very small amounts of N‑glycans, to a glycoprotein biosimilar. Our ability to provide direct evidence of the expected, modified glycan structure was crucial to the funding prospects of the company developing the therapy
 |
Protein Sequencing
Protein Sequencing
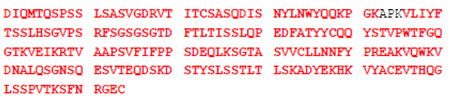
A biosimilar is a copy version of an already authorized biological medicinal product with demonstrated similarity in physicochemical characteristics, efficacy, and safety. Comprehensive physicochemical characterisation of (glyco)protein biosimilars includes determination of amino acid sequence, usually by mass spectrometry. We are able to provide high sequence coverage of proteins such as immunoglobulins using a combination of peptide fingerprinting and T3 sequencing, we can sequence well over 90% of a many proteins. The example shown here is Avastin® (bevacizumab), a recombinant humanized monoclonal antibody that is used to treat various cancers. Only the amino acids shown in black were not detected.
Light Chain

Heavy Chain