Professor Peter Karadakov
Emeritus Professor
Email: peter.karadakov@york.ac.uk
My research interests are in the area of molecular electronic structure theory and involve methodological work and code development, as well as theoretical studies of bonding, aromaticity, reactivity and other electronic properties of molecular systems in organic, inorganic chemistry and materials science. Here are several topics in which we have been interested in recent years.
Predicting Excited State Aromaticity Reversals Using Magnetic Criteria
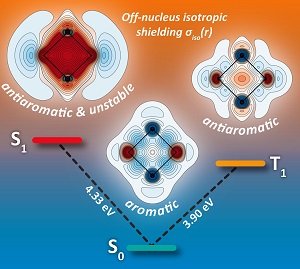
Surprisingly, it is often possible to reverse the aromaticity of a molecule by exciting it to a different electronic state, either from aromatic to antiaromatic, or in the opposite direction, from antiaromatic to aromatic. Such aromaticity reversals are accompanied by profound changes in molecular electronic structure and properties. Understanding this type of molecular behaviour can help in the design of molecular photoswitches and molecular motors and explain experimental evidence for photochemical reactions such as excited-state intramolecular proton transfers.
We have shown that magnetic properties such as nucleus-independent chemical shifts (NICS), proton shieldings, magnetizabilities and isotropic magnetic shielding contour plots and isosurfaces, calculated using state-optimized complete-active-space self-consistent field (CASSCF) wavefunctions, provide robust theoretical tools for identifying ground and excited state aromaticity and antiaromaticity. The strikingly different shielding distributions in the ground states of benzene and cyclobutadiene can be viewed as aromaticity and antiaromaticity “fingerprints” that are reproduced in other electronic states of these and other molecules and allow the classification of various electronic states as aromatic or antiaromatic. Isotropic magnetic shielding contour plots and isosurfaces, the method that we have been using, compute essentially the same magnetic shielding that influences chemical shift in nuclear magnetic resonance spectroscopy. But while NMR shifts are centred on an atom, IMS indicates what is happening between atoms, and hence provides insight into the bonding in molecules. The only drawback is that to obtain all this information, we need to do a lot of computational work.
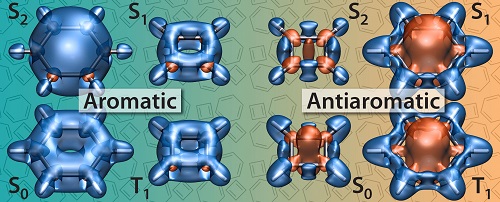
Recent results indicate that the very similar levels of antiaromaticity in the S1 and T1 states of benzene, and of aromaticity in the S1 and T1 states of cyclobutadiene are exceptions rather than the rule: We have established important differences between the antiaromatic S1 and T1 states of disulfur dinitride and even more pronounced differences between the S1 and T1 states of simple polycyclic aromatic hydrocarbons (PAHs) such as naphthalene and anthracene which indicate that singlet and triplet (or “Baird”) excited state aromaticities are different and largely independent phenomena which need to be studied separately.
P. B. Karadakov, Ground and Excited State Aromaticity and Antiaromaticity in Benzene and Cyclobutadiene, J. Phys. Chem. A 112 (2008) 7303–7309.
P. B. Karadakov, Aromaticity and Antiaromaticity in the Low-Lying Electronic States of Cyclooctatetraene, J. Phys. Chem. A 112 (2008) 12707-12713.
P. B. Karadakov, P. Hearnshaw and K. E. Horner, Magnetic Shielding, Aromaticity, Antiaromaticityand Bonding in the Low-Lying Electronic States of Benzene and Cyclobutadiene, J. Org. Chem. 81 (2016) 11346-11352.
P. B. Karadakov, M. A. H. Al-Yassiri and D. L. Cooper, Magnetic Shielding, Aromaticity, Antiaromaticity and Bonding in the Low-Lying Electronic States of S2N2, Chem. Eur. J. 24(2018) 16791-16803.
B. J. Lampkin, Y. H. Nguyen, P. B. Karadakov and B. VanVeller, Demonstration of Baird's rule complementarity in the singlet state with implications for excited-state intramolecular proton transfer, Phys. Chem. Chem. Phys. 21 (2019) 11608-11614.
P. B. Karadakov and S. Saito, Can Anti-Aufbau DFT Calculations Estimate Singlet Excited State Aromaticity?, Angew. Chem. Int. Ed. 59 (2020) 9228-9230.
P. B. Karadakov and D. L. Cooper, Excited-state Aromaticity Reversals in Möbius Annulenes,
J. Phys. Chem. A 124 (2020) 9611-9616.
R. Kotani, L. Liu, P. Kumar, H. Kuramochi, T. Tahara, P. Liu, A. Osuka, P. B. Karadakov and S. Saito, Controlling the S1 Energy Profile by Tuning Excited-State Aromaticity, J. Am. Chem. Soc. 142 (2020) 14985-14992.
Making Aromaticity ‘Klar’
The computational methodology developed for studying excited state aromaticity reversals provides an easy way of visualising the aromaticity of important polycyclic aromatic hydrocarbon molecules and gives new insights into their bonding.
In the 1960s, Erich Clar developed his sextet rule, which states that a molecule’s dominant resonance structure is the one with the largest number of disjointed aromatic π-electron sextets. Clar sextets can be depicted by a circle drawn inside a benzene hexagon. Yet, there is still a lot of confusion around Clar’s rule, with many people simply incorrectly drawing a circle in all of the rings with any aromatic character.
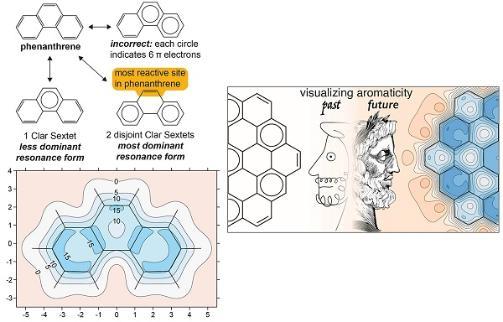
(Top left) Structure of phenanthrene drawn in the traditional way showing Clar sextets both correctly and incorrectly. (Bottom left) The magnetic shielding map provides an intuitive way to understand aromaticity – in this case phenanthrene is shown and the darker blue indicates greater aromaticity, indicating that the terminal rings are more aromatic than the central one. (Right) Past and future in visualizing aromaticity.
Our isotropic magnetic shielding plots provide an intuitive picture of aromaticity – both overall and local. These pictures are remarkably powerful - as soon as you see them, you can immediately start interpreting them. In phenanthrene, for example (see image), the picture clearly shows that the terminal rings are darker in colour, therefore more aromatic, than the central ring – as is predicted by Clar’s rule. Our approach can replicate Clar’s rule when it is accurate, but importantly, we can go beyond it when it’s inaccurate. For example, the molecule coronene cannot be drawn with a single static representation that encompasses all of its resonance forms. But the new contour plots showed immediately, at a glance, that all of coronene’s outer rings are equally aromatic.
B. J. Lampkin, P. B. Karadakov and B. VanVeller, Detailed Visualization of Aromaticity Using Isotropic Magnetic Shielding, Angew. Chem. Int. Ed. 59 (2020) 19275-19281.
Modern Valence-Bond Theory
The ability of a modern form of valence-bond (VB) theory, the spin-coupled generalized valence bond (SCGVB) method, to include a significant amount of electron correlation within a wavefunction, which remains easy-to-visualize and interpret, provides us with a powerful tool for studying not just bonding in isolated molecules, but also the bond-breaking and bond-formation processes that take place during chemical reactions.
In its original form the SCGVB wavefunction makes use of a “N in N” active space, described by means of a single product of N non-orthogonal active orbitals, multiplied by a general N-electron spin function. The “N in M” active spaces with different numbers of active electrons and active orbitals which are encountered in many CASSCF applications can be handled through our extension of SCGVB theory to “N in M” (N ≠ M) active spaces.
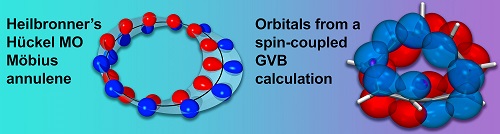
We have shown that the fully-variational optimization of an ab initio “N in M” SCGVB wavefunction vindicates, in a surprising level of detail, essential features of Heilbronner’s ideas for the electronic structure of Möbius annulenes such as the arrangement of overlapping carbon 2p atomic orbitals along a Möbius strip, leading to a phase inversion between the first and last orbitals. In the SCGVB description, the aromaticity of this Möbius system follows from the extensive resonance between VB structures.
P. B. Karadakov, D. L. Cooper, B. J. Duke and J. Li, Spin-Coupled Theory for “N in M” Active Spaces, J. Phys. Chem. A 116 (2012) 7238-7244.
P. B. Karadakov and D. L. Cooper, Does the Electronic Structure of Möbius Annulenes Follow Heilbronner's Ideas?, ChemPhysChem 19 (2018) 3186-3190.

