Dr Martin Cockett
01904 324534
E-mail: martin.cockett@york.ac.uk
High Resolution Laser Spectroscopy with Ions and Electrons
Our research interests focus on experimental and computational study of a wide range of molecular systems, with a particular emphasis on non-covalent interactions in molecular complexes. Our experimental methodology of choice is gas phase electronic laser spectroscopy which we combine with state-of-the-art computational chemistry to elucidate the conformations adopted by molecules in the gas phase and to determine how these conformations change in response to changes in electronic state. In the context of our interests in van der Waals complexes, our aim is to identify the most energetically favoured conformation in each electronic state involved in an electronic transition and to ascertain the extent to which the stability of each conformer depends on the electronic environment of the chromophore: such considerations have particular resonance in the fields of molecular association and recognition.
Van der Waals complexes: Synthesis and Study
Van der Waals interactions are important in a number of areas in chemistry: for example they play a vital role in determining the selectivity and function of biologically active molecules; they define the process by which gases condense to liquids; and ionic clusters play an important role in ion-molecule chemistry in flames and plasmas, in the terrestrial atmosphere and in interstellar space. We synthesise van der Waals complexes in the gas phase using supersonic free jet expansion techniques and then study them using electronic laser spectroscopy. Mass resolved REMPI (resonance enhanced multiphoton ionisation) spectroscopy is used to study electronically excited states of the neutral, and both ZEKE-PFI (zero kinetic energy - pulsed field ionisation) and MATI (mass analysed threshold ionisation) spectroscopy are used to study the cation. Experimental spectra are interpreted with the aid of high level ab initio calculations of ground and excited states in conjunction with multidimensional Franck-Condon analysis.
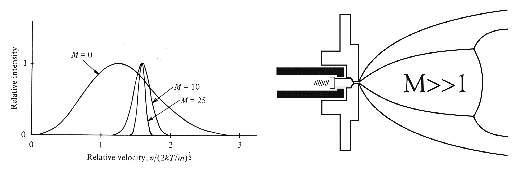
Figure 1. Expansion of a gas through a small aperture with a sufficiently high pressure differential results in adiabatic cooling and a narrowing of the internal energy distribution of the gas molecules that increases with increasing Mach number. The cooling achieved in such a supersonic jet expansion is sufficient to allow weakly bound van der Waals complexes to be formed in the jet.
These exotic molecules are characterised by very low binding energies, very long bond lengths and very low frequency vibrational motions. Consequently, they present a rather unique challenge to the experimentalist as well as to the theoretician. Our experiments allow us to characterise the so-called intermolecular vibrational modes in electronically excited states of the neutral molecule as well as in the ground state of the ion. We can also deduce the structure of the complex and the extent to which solvation affects the potentials describing the conformation and intramolecular bonding in the solute. We can observe not only the effect of solvation on the solute species but also the effects of changes in the electronic environment of the species on the van der Waals interaction. Some of the questions we hope to address include: To what extent does the solvent affect the intramolecular modes of the solute? Can we use vibrational wavenumber shifts upon electronic excitation as a fingerprint of the molecular conformation? How does the van der Waals interaction change if we excite the molecule into a valence or Rydberg excited state? What happens if we ionise one component of the complex?
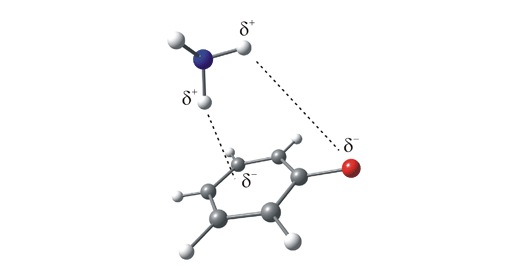
Figure 2. One possible conformation of the fluorobenzene-ammonia complex. This is an example of a complex in which the ammonia (the solvent) binds to the solute (fluorobenzene) via two weak interactions: one, a weak π-hydrogen bond formed between a δ+ hydrogen on the ammonia and the π-system on the aromatic ring, and the other between a second ammonia hydrogen and the electron rich fluorine substituent.
The weak hydrogen bond in amino-aromatic complexes
The most ubiquitous type of non-covalent interaction is the hydrogen bond, the strength of which is wide-ranging, extending from the weakest CH- and NH- π interactions in molecules containing aromatic rings to much stronger, more directional X-H···Y interactions where X and Y are highly electronegative atoms. At the weaker end of this scale, π-hydrogen bonds have been recognised in the past few years for the important role they play in molecular recognition and association, molecular biology and crystal engineering. In the context of molecular biology, they have been shown to feature not only in the stabilisation of protein secondary structures, but also in the binding of ligands in proteins. In spite of their weaker nature, π-hydrogen bonds in aromatic ligands are pivotal to protein–ligand recognition, yet far less is known about what determines their strength and in general about how the relative strengths of the different types of interaction contribute globally to the binding constant of a ligand within a protein cavity. Our work aims to address this short-coming by combining advanced laser spectroscopic techniques with newly developing ab initio methodologies and state-of-the-art vibrational analysis in a systematic study of the effect of substituent choice on the stability of different types of weak hydrogen bond in model systems composed of an aromatic or heteroaromatic chromophore and an amine. The model complexes chosen mimic key structural components of small aromatic ligands commonly used in protein inhibitors. Our hope is that this work will provide the basis of a much better understanding of, in particular, NH-π interactions, with the exciting potential for longer term exploitation in molecular recognition, and more specifically in structure-based design of improved binding affinity ligands.
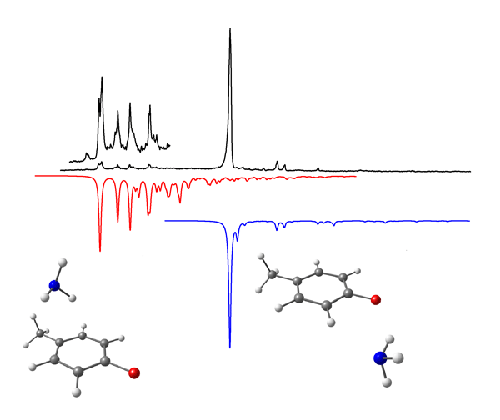
Figure 3. The presence of an electron-donating methyl group in p-fluorotoluene-ammonia stabilises the π-complex to the extent that bands associated with two conformers are seen in the REMPI spectrum. The top spectrum is the experimental spectrum and the red and blue spectra (lower, inverted) are the result of Franck-Condon simulations obtained for each conformer (see below).
Multidimensional Franck-Condon Analysis of Electronic Spectra
FC-LabII
The vibrational activity seen in an electronic spectrum contains detailed information relating to the extent to which the molecule has undergone changes in its conformation as a consequence of its change in electronic state. Geometry changes along a particular internal coordinate will generally result in activity in a vibrational modes that reflect the extent of that change along the coordinate. For example, in a simple diatomic molecule, excitation of an electron from a bonding orbital to an anti-binding orbital will weaken the bond strength in the excited state resulting in a longer equilibrium bond length. A electronic spectrum that probes such a change in state would then be expected to show a vibrational progression in the stretching mode. In a non-linear polyatomic molecule, there are 3N-6 vibrational degrees of freedom and we can necessarily expect that the vibrational structure observed in an electronic spectrum will be considerably more complex than for a diatomic molecule, reflecting changes in geometry along numerous internal coordinates. The interpretation of such spectra can sometimes be achieved using good old fashioned chemical intuition, but in the majority of cases, successful vibrational analysis and assignment can only be achieved with the aid of detailed molecular orbital calculations combined with so-called multidimensional Franck-Condon analysis.
 .
.
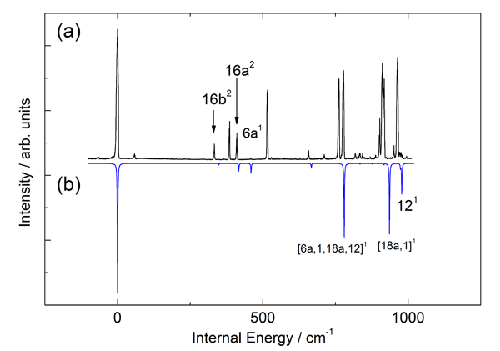
Figure 4. A comparison of (a) the experimental REMPI spectrum of fluorobenzene with (b) a Franck-Condon simulation based on the results of ab initio calculations conducted at the RICC2/def2-TZVPP level of theory. This comparison allows an assignment of the key vibrational features of the REMPI spectrum whilst also highlighting the significant extent to which vibronic coupling lends intensity to bands in the experimental spectrum whose appearance is formally forbidden.
This project involves a collaboration with Drs. Igor Pugliesi and Christian Schriever at the Ludwig-Maximilians-Universität in Munich to develop a programme that allows simulation of electronic spectra through the computation of Franck-Condon factors for each of the vibrational degrees of freedom in the molecule. View details about the programme.
Rydberg excited van der Waals complexes
Rydberg states of atoms and molecules are unusual because one electron occupies an orbital whose size is large relative to the dimensions of the ionic core. We are interested in investigating Rydberg states of van der Waals complexes which exhibit bonding characteristics defined largely by whether or not the cluster notices the Rydberg electron. This depends largely on how excited the electron is (i.e. the value of the principal quantum number) and the extent to which it penetrates the region close to the ionic core of the molecule (given by the value of the orbital angular momentum quantum number, l). We have studied complex Rydberg states whose value of n ranges from just 3 to somewhere in the region of 200. One of the key question in this work is whether or not the dynamic properties of the core (such as vibration and rotation) can interact with the dynamic properties of the electron. We can also get a sense in low-lying Rydberg states of the relationship between the solvent and the Rydberg electron by whether or not the complex is stabilised or destabilised by electronic excitation relative to the neutral ground state. Recent examples of our work in this area include the observation of Divergent energy level spacings as evidence for an offset parallel structure in the DABCO-N2 complex (DABCO = 1,4-diazabicyclo[2,2,2]octane); The role of symmetry and optical selection rules in revealing the molecular structure of the DABCO-Arn (n=1,2,3) complexes and perhaps the most exciting for us, the observation of well resolved, long-lived (>10 μs) hydrogenic Rydberg states in the range n=50 to 90 in the DABCO-N2 complex. This is the largest system displaying such characteristics ever reported.
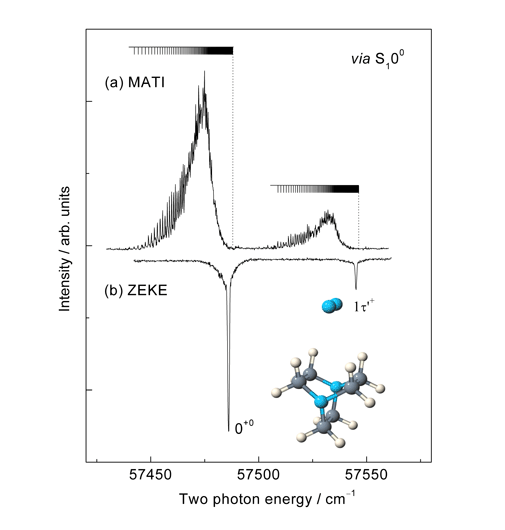
Figure 5. A comparison of the (a) MATI and (b) ZEKE spectra of the DABCO-N2 complex, each recorded via the S1 band origin. Each spectrum shows a short progression in a van der Waals vibrational mode but in addition the MATI spectrum show additional fine structure associated a single Rydberg series extending from about n=50 to the respective vibronic thresholds.

