Dr Han-Jou Chen
Lecturer in Neuroscience
Research

Why do proteins aggregate?
As the ageing population increases, age-dependent neurodegenerative diseases such as Amyotrophic Lateral Sclerosis (ALS), Alzheimer’s Disease (AD), Frontotemporal Dementia (FTD) and Parkinson’s Disease (PD) cause enormous emotional, medical and financial impact on both patients and the society. These diseases are caused by the progressive neuronal loss which eventually results in impairment of the motor and/or cognitive function of the affected individuals. Sadly, for most neurodegenerative conditions, there is currently no effective therapy.
Studies into neurodegenerative disorders have identified that despite different neurons are affected in different diseases, most neurodegenerative diseases show a common feature that is the accumulation of protein aggregates in affected cells, such as TDP-43 in ALS, Aβ and tau for AD, α-synuclein in PD, and TDP43 or tau in FTD. These aggregated proteins often bear abnormal post-translational modification such as phosphorylation, and they also transform from their native structure to adapt a more rigid, detergent-resistant structure that is prone to further aggregation. Evidence shown that these aberrant protein aggregates are linked to cytotoxicity and neuronal loss. To understand why these protein aggregates and providing a therapeutic solution, our research focus on 1) investigating the underlying mechanisms that contribute to protein aggregation, and 2) explore the potential of chaperone response system as therapy for neurodegenerative diseases.
To maintain cellular protein homeostasis, cells express multiple chaperone proteins to facilitate the folding and degradation of aberrant proteins. We focus on a group of chaperones called heat shock proteins (HSPs) which is comprised of chaperones such as HSP90 and HSP70 families, co-chaperones such as HSP40 and HSP110 families and small HSPs. Some HSPs such as HSP90 and HSP70 are maintained at a steady level in most cells, but other HSPs, especially the HSP40s, are kept at a low endogenous level which are induced during cellular stress. The induction of HSPs are carried out by the transcription faction HSF1. During cellular stresses, HSF1 is activated which in turn activates the expression of downstream HSPs. Our study shows that the aggregation and cytotoxicity of TDP-43 is rescued by HSP70/DNAJB2a induced by HSF1. Interestingly, the levels of HSF and HSPs are significantly lower in affected tissues of ALS patients and animal model, implying the reduced activity of chaperone response may contribute to the accumulation of aberrant TDP-43 and the disease development.
Apart from chaperone proteins, we also investigate the role of RNA in protein aggregation. Our recent study shows that when being prevented to interact with RNA, TDP-43 proteins are more prone to form hyperphosphorylated and detergent-resistant aggregates. On the other hand, mixing RNA with purified TDP-43 peptide delays the aggregate formation in the test tube. These results indicate as a DNA/RNA binding protein, the interaction between TDP-43 protein and its target RNA has kept TDP-43 in its native soluble structure. Any disruption on this interaction will lead to TDP-43 aggregation. However, the exact mechanism involved in this process required further clarification
Finally, to translate our finding into clinical impact, we are currently exploring the potential of targeting HSF1 for ALS treatment. Support by the Motor Neurone Disease Association, we are testing whether direct activation of HSF1 in neurones can remedy the protein aggregation, neuronal loss and motor defects of a TDP-43 mouse model. Collaborating with a Drug Discovery Institute, we are also screening for new drugs to activate HSF1 and provide a new therapeutic option for ALS.
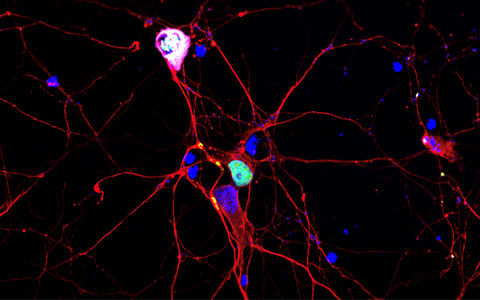
TDP-43 protein aggregates in primary neurons. B3-tubulin is used as a neuronal marker stained in red. TDP-43 (green) is localised at the nucleus under normal condition and can be seen to form aggregates along the neurites. Aberrant TDP-43 is further phosphorylated (magenta) and aggregated in the cytoplasm.
Teaching and scholarship
![]()
I believe teaching is not only about passing down the knowledge, but inspiring a way of thinking, an attitude. Based on this belief, I aim to present the most up-to-date research results to the lecture and stimulate critical thinking.
![]()
I am currently teaching year 3 Neuroscience and Genetics. My neuroscience lectures focus on the disease affecting the CNS, such as ALS. In these lectures, I will cover the profile of the diseases, what goes wrong, and what the current understanding of the diseases causing mechanisms are. Genetics often provides fundamental knowledge to understand complex biological process and system. It is also a powerful tool in the development of disease modelling and treatment. My lectures in Human Genetics will cover the commonly used genetic modelling organisms, technologies involved in creating them and discussions on gene therapy.
![]()
I run neuroscience tutorials where students can choose any subject they are interested in within neuroscience. Together, we will discuss the subject selected, read and discuss research papers in the field and put together an essay presenting comprehensive argument regarding our understanding (or lack of understanding) on this subject.
![]()
Projects aligned with my research interest are offered to students keen to research in neurodegeneration. These will be cell-based study investigating the contributing factors of protein aggregation.
